
電話:
021-67610176傳真:
來自德克薩斯大學西南醫學中心,QBRC、BICF中心主任謝陽教授實驗室在QB期刊上發表關于分析MeRIP-Seq (methylated RNA immunoprecipitation sequencing)數據的新方法(A Bayesian hierarchical model for analyzing methylated RNA immunoprecipitation sequencing data)。

在《A Bayesian hierarchical model for analyzing methylated RNA immunoprecipitation sequencing data》這篇文章中,我們提出用一種貝葉斯統計模型,即貝葉斯層次模型BaySeqPeak,用于分析MeRIP-Seq數據,從而幫助研究人員發現轉錄組中的甲基化位點信號[1]。
RNA甲基化數據分析現狀
DNA與組蛋白的表觀遺傳修飾在調控基因表達上的重要影響已為科學界所廣泛熟知。同DNA一樣,作為生物遺傳信息傳遞中的重要一環,RNA分子也廣泛存在著化學修飾。目前,科學家已經鑒定確認了超過100種的RNA化學修飾方式,其中以m6A(N6-methyladenosine,6-甲基腺嘌呤,化學結構見圖1)為常見[2]。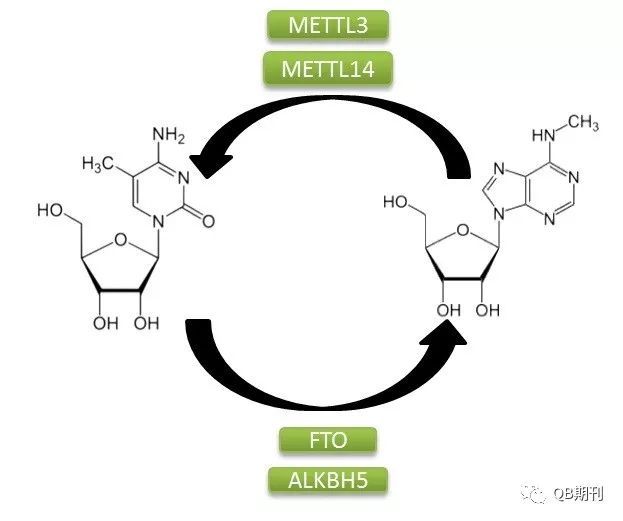
圖1 m6A甲基化修飾過程
m6A甲基化修飾是一種由多種蛋白參與的動態可逆的修飾方式。它的生成主要是由甲基轉移酶復合體介導,其中包含METTL3,METTL14和WTAP;而擦除甲基化修飾基團的過程則由去甲基化酶FTO和ALKBH5負責。此外,多種蛋白,如YTHDF1和YTHDF3都可識別m6A信號位點,并通過結合下游效應蛋白的方式傳遞甲基化信號。目前已經發現,m6A在調控基因表達、剪接、RNA 編輯、RNA 穩定性和控制mRNA壽命和降解等多方面都存在重要的影響[3]。
雖然RNA甲基化在上世紀七十年代就已經被發現證實,但長期以來由于技術局限,相關的修飾機理、調控手段以及生物學意義一直未能闡明。現在,MeRIP-Seq (methylated RNA immunoprecipitation sequencing)技術的出現[4,5](圖2),使得通過高通量手段在全轉錄組(transcriptome)水平上研究m6A甲基化修飾變為可能。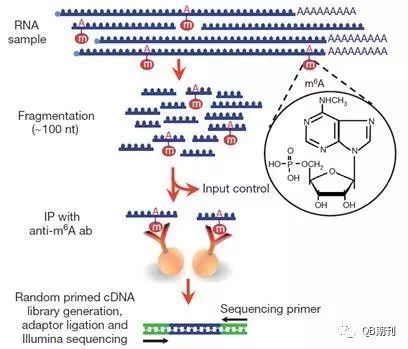
圖2 MeRIP-Seq技術流程圖
通行的分析MeRIP-Seq數據的思路是,利用一個特定長度的(通常為100~200nt長)窗口從前至后掃描整條染色體,并記錄每個樣本落入每個窗口中的RNA短序列數目(read count)。通常,實驗條件下(IP)樣本的RNA短序列應大致分布在甲基化位點附近,而對照條件下(INPUT)則和正常的每個基因的表達值正相關(沒有甲基化影響)。這一數據特點使得傳統分析DNA甲基化的工具無法很好地勝任RNA甲基化數據的分析。總而言之,轉化為統計語言就是,我們需要尋找那些在實驗條件下序列數目顯著高于對照條件下序列數目的窗口(甲基化位點),并相應地給予顯著性統計值(p值或假陽性概率)。
BaySeqPeak模型分析RNA甲基化數據的優勢
我們建立的BaySeqPeak模型則主要從MeRIP-Seq數據的重復樣本數少,樣本數據空間上前后相關,以及存在大量零數據的特點出發,利用以下三種不同的策略解決了這些難題:
1)采用零膨脹的負二項分布擬合單樣本的序列計數,以防止大量零數據和過度離散破壞模型穩定性;
2)采用隱馬爾可夫模型模擬單樣本空間上的前后相關性;
3)利用貝葉斯統計的思路,使得模型在低樣本數的條件下依然維持足夠的準確度。
在模擬數據中,BaySeqPeak能很好地預測了實驗人員預先設定的甲基化位點,而比較的exomePeak和MeTPeak模型則匯報了較多的假陽性和假陰性位點(圖3)。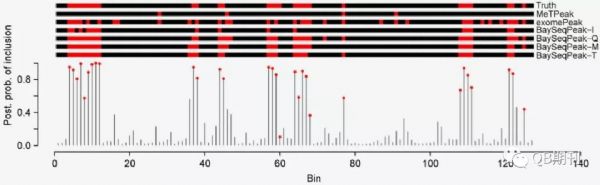
圖3 模擬數據中真實的甲基化位點與各模型預測的甲基化位點(紅色)
通過ROC曲線可以發現,不同參數下的模擬數據中,BaySeqPeak模型的預測準確性均顯著高于exomePeak和MetPeak模型(圖4)。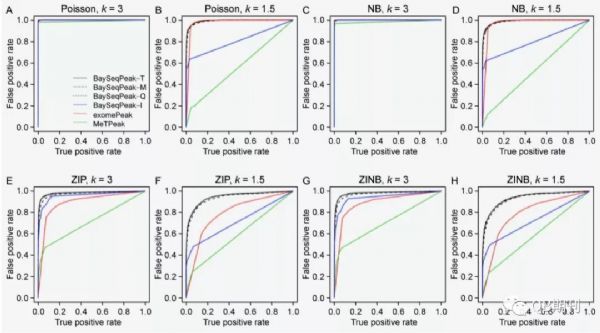
圖4 不同參數條件下,各模型預測的ROC曲線
在數值收斂方面,模型在經過多次迭代之后,預測值已穩定地收斂到了真實值附近(圖5)。
圖5 甲基化位點的預測數的收斂過程
在真實數據中,BaySeqPeak模型也很好地預測了甲基化的區段。不僅如此,相較于exomePeak,BaySeqPeak還詳細區分出了一個甲基化區域中臨近的幾個甲基化峰位,顯示了模型的高精度與高分辨率。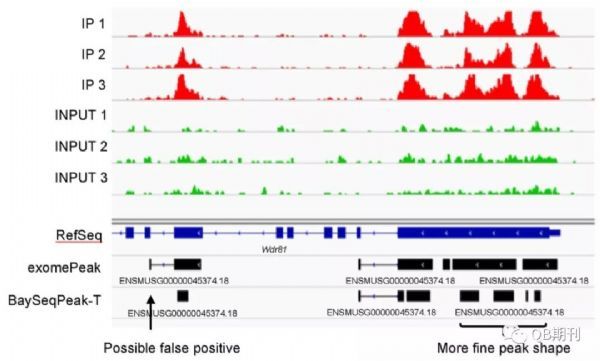
圖6 一個真實數據中預測的甲基化區域
RNA甲基化的研究目前仍然處于起步階段,修飾調控過程的具體細節,以及這些修飾如何具體地影響細胞的功能,特別是在疾病條件下,這些化學修飾是如何發生變化的仍然存在大量未知。本文提出的統計方法為有效準確地分析m6A甲基化數據提供了可能,我們期待在未來RNA甲基化的研究中能夠在此模型基礎上再推進一步。
參考文獻
Zhang, M., Li, Q., & Xie,Y. (2018). A Bayesian hierarchical model for analyzing methylated RNA immunoprecipitationsequencing data. Quantitative Biology, 6(3), 275-286.
Machnicka, M. A., Milanowska,K., Oglou, O., Purta, E., Kurkowska, M., Olchowik, A., Januszewski, W.,Kalinowski, S., Dunin-Horkawicz, S., Rother, K. M., et al. (2013) MODOMICS: adatabase of RNA modification pathways–2013 update. Nucleic Acids Res., 41,D262–D267
Meyer, K. D., and Jaffrey, S.R. (2014) The dynamic epitranscriptome: N 6-methyladenosine and gene expressioncontrol. Nat. Rev. Mol. Cell Bio., 15, 313–326
Dominissini, D.,Moshitch-Moshkovitz, S., Schwartz, S., Salmon-Divon, M., Ungar, L., Osenberg,S., Cesarkas, K., Jacob-Hirsch, J., Amariglio, N., Kupiec, M., et al. (2012)Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature485, 201–206.
Meyer, K. D., Saletore, Y.,Zumbo, P., Elemento, O., Mason, C. E. and Jaffrey, S. R. (2012) Comprehensiveanalysis of mRNA methylation reveals enrichment in 3′ UTRs and near stopcodons. 1517 Cell, 149, 1635–1646.
13224517959

